Research
My interests lie at the interface of technology with (bio)pharma and healthcare. In the past, I developed and applied machine learning and physics-based methods for drug, protein, and polymer design; below are some examples.
Molecular property prediction
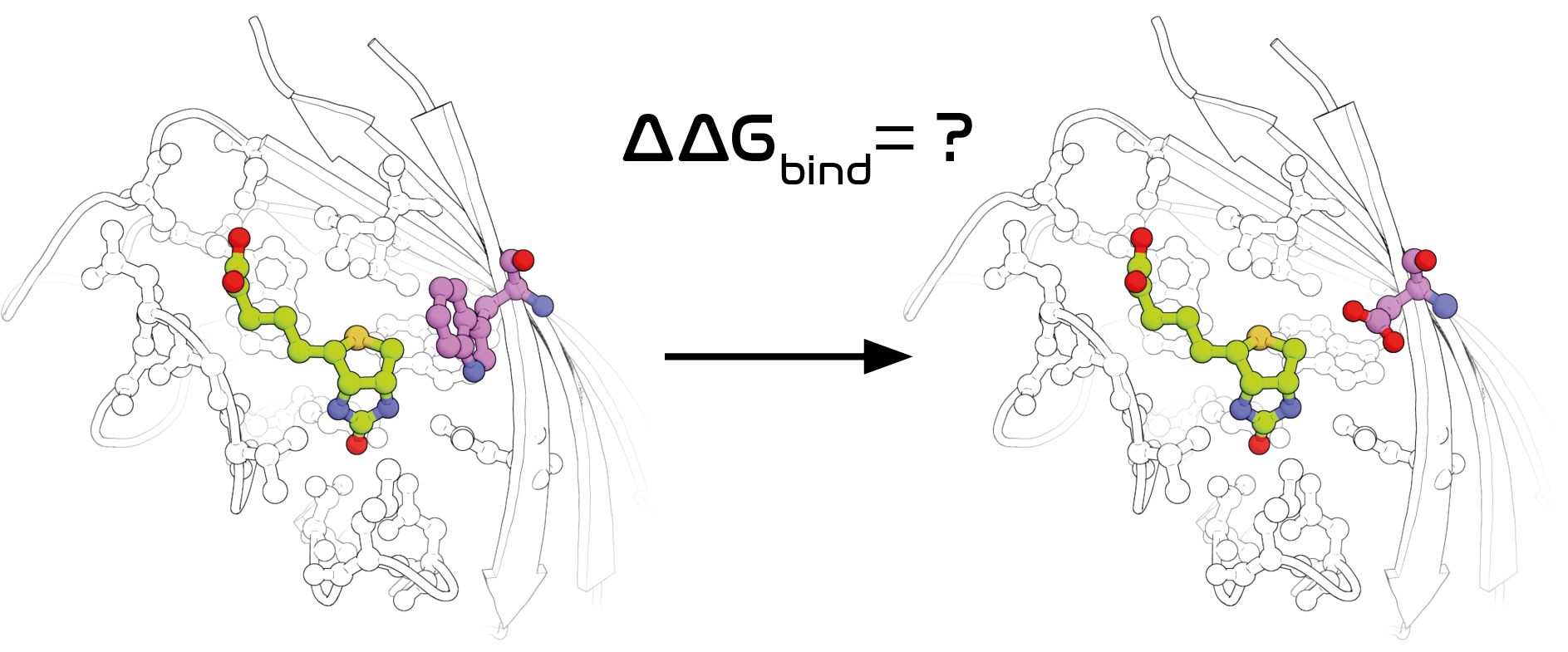 Leveraging computation for the design of organic and biological molecules often means trying to predict (bio)molecular properties. I've worked on predicting protein-ligand binding affinity with molecular dynamics simulations (Chem. Sci.
Leveraging computation for the design of organic and biological molecules often means trying to predict (bio)molecular properties. I've worked on predicting protein-ligand binding affinity with molecular dynamics simulations (Chem. Sci.
Active learning and design of experiment
 Numerous tasks in science and engineering can be framed as multi-objective optimization problems. I've spent some time working on software and algorithms for model-guided optimization (Acc. Chem. Res.
Numerous tasks in science and engineering can be framed as multi-objective optimization problems. I've spent some time working on software and algorithms for model-guided optimization (Acc. Chem. Res.
Biomolecular mechanisms and dynamics
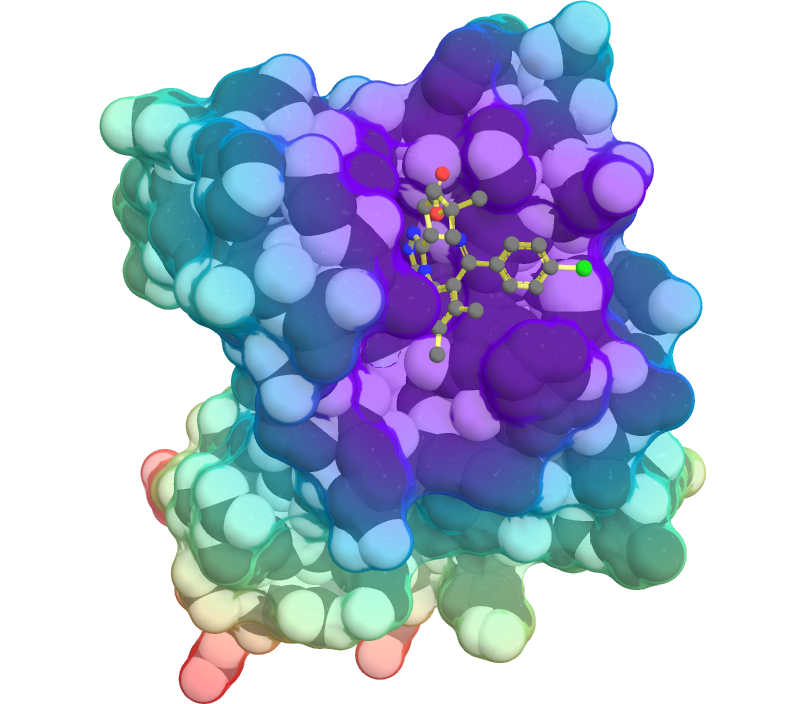 Computer simulations allow one to study biochemical systems in atomistic detail, exploring the multitude of conformations available to molecules in solution, and extracting information about their thermodynamic, kinetic, and dynamical properties. Ultimately, they provide insight into the molecular determinants of biological function and dysfunction. In the past, I've used simulations to study the role of water molecules in mediating protein-ligand binding (Sci. Adv.
Computer simulations allow one to study biochemical systems in atomistic detail, exploring the multitude of conformations available to molecules in solution, and extracting information about their thermodynamic, kinetic, and dynamical properties. Ultimately, they provide insight into the molecular determinants of biological function and dysfunction. In the past, I've used simulations to study the role of water molecules in mediating protein-ligand binding (Sci. Adv.